Photon Counting Detectors
High-performance SPAD, PMT and hybrid detectors for reliable photon counting across materials science, life science, quantum optics, and metrology.


Simplify your materials characterization with one flexible TRPL microscope enabling multiple methods for precise and efficient analysis.

Complete confocal fluorescence microscope that empowers researchers to advance quantitative functional imaging from individual molecules to cells and tissues.

Compact FLIM and FCS upgrade kit that adds advanced functional imaging and correlation analysis to existing laser scanning microscopes.

Designed for flexible, sensitive, and precise steady-state and time-resolved spectroscopy across the UV to NIR range and time scales from picoseconds to milliseconds.

Modular lifetime spectrometer designed for flexible fluorescence and photoluminescence measurements in both materials and life science research.

Add spectral and time-resolved photoluminescence to your setup through flexible microscope–spectrometer coupling options.

Get the most out of superconducting nanowire detectors in large-scale quantum communication and computing experiments requiring precise multichannel timing.

Boost your time-resolved experiments with a flexible, high-precision time tagging and TCSPC unit for materials science and quantum sensing.

Scale your photonic quantum computing and detector characterization setups while maintaining performance, flexibility, and high data throughput.

Compact 3-color picosecond laser delivering flexible ns to ms excitation with cost-effective multicolor performance and straightforward operation.

Smart picosecond laser diode heads covering UV-A to NIR, providing the right combination of power, pulse width, and diode type for any time-resolved technique.

VisUV provides clean short pulses and stable timing across key UV and visible wavelengths, including deep UV lines as well as 488 nm and 532 nm.

Enhance your single-photon counting experiments with wide dynamic range and excellent timing precision in the UV and visible even at the highest count rates.

Capture even the weakest signals over large areas with maximum dynamic range and enhanced low-light sensitivity in a compact detector design.

Unlock spatially resolved single-photon detection with a 23-pixel SPAD array, combining low dark counts and precise time tagging for advanced experiments.
Advanced FLIM analysis software for fast, accurate interpretation of lifetime imaging data.
Intuitive, free software solution for real-time, high-precision photon data acquisition, visualization, and initial data analysis.
Advanced software for time-resolved fluorescence acquisition and analysis.
An imaging technique that uses fluorescence lifetimes to generate image contrast.
Investigating how proteins dynamically explore multiple conformational states that control biological function.
Investigating how biomolecules separate into dynamic liquid phases to organize cellular space and regulate biological function.
A time-resolved technique that measures photoluminescence lifetimes to reveal excited-state dynamics in materials.
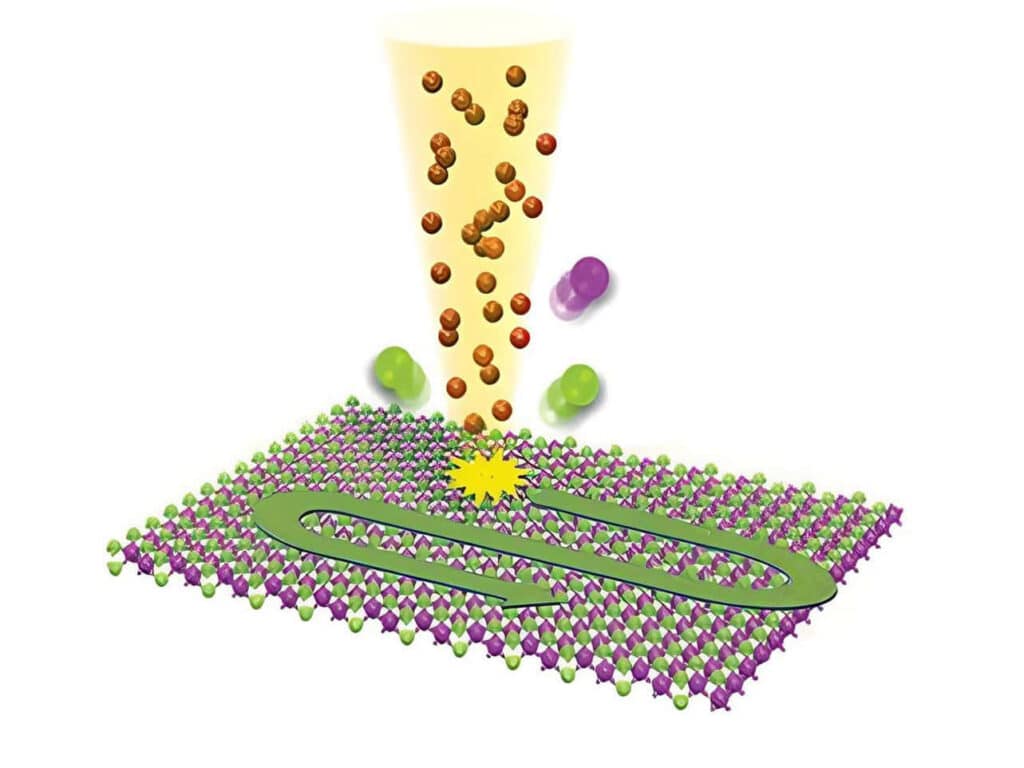
Studying exciton dynamics, charge carrier processes, and structural properties through optical and time-resolved characterization methods.

Investigating charge-carrier lifetimes and recombination dynamics to enable precise optical characterization of material quality and device performance.
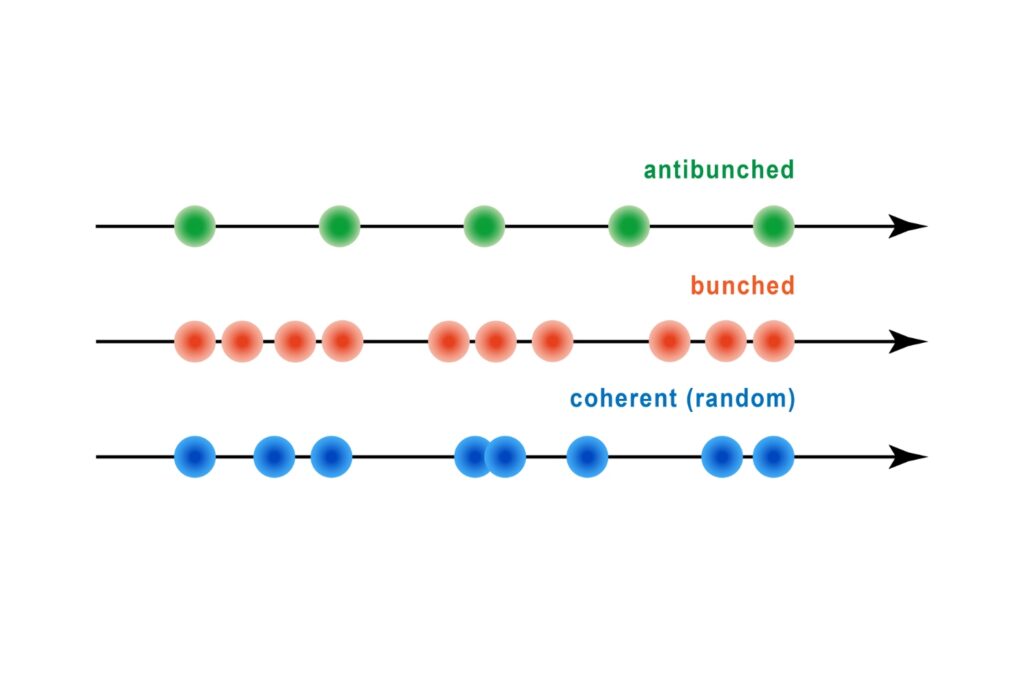
A quantum optical signature revealed by time-resolved photon correlation analysis to identify single-photon emission in materials and nanostructures.
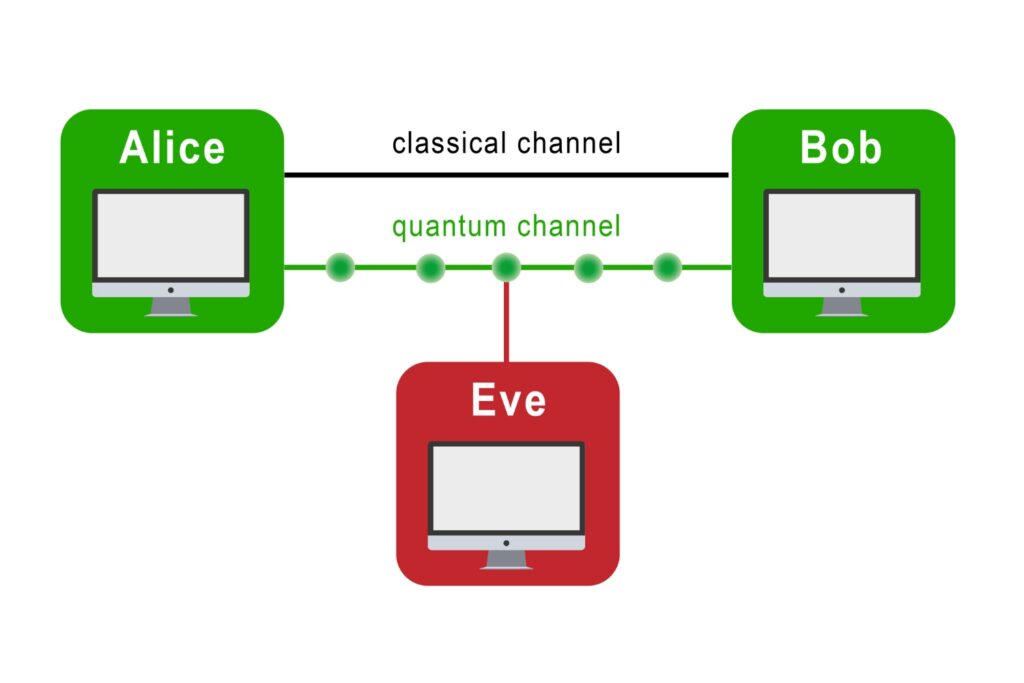
The transmission of information using individual photons, using quantum effects to ensure absolute security.
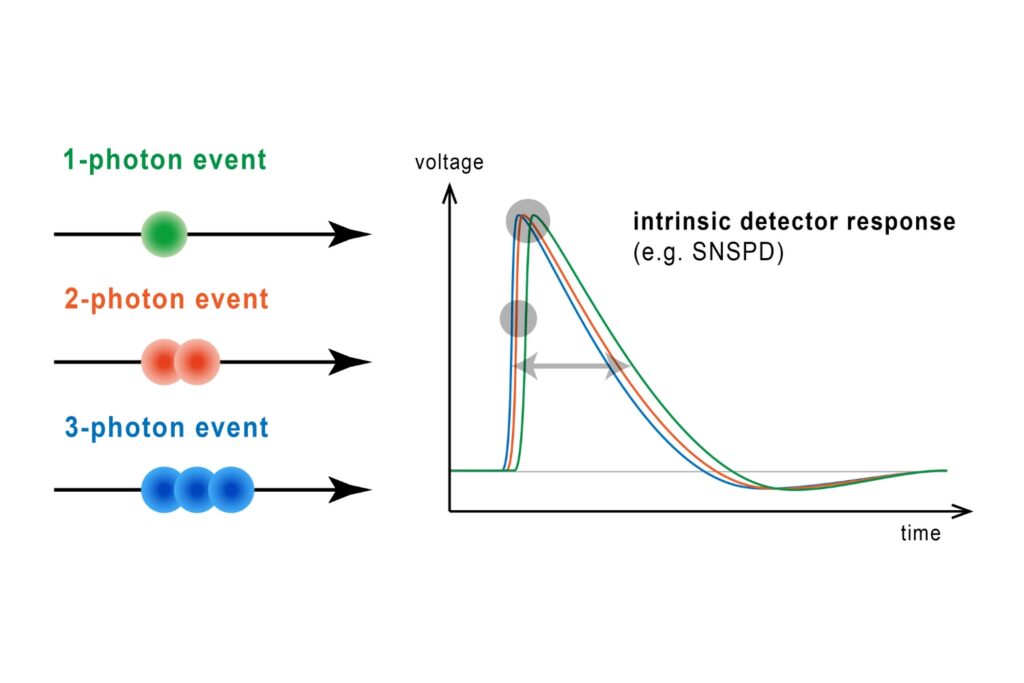
Quantifying photons per detection event enables direct access to photon-number statistics, providing insight into quantum and statistical properties of light.

An optical technique that analyzes light emission under electrical excitation to reveal electronic properties of electroluminescent materials.
Monitoring environmental signals and trace compounds to understand dynamic changes in natural and engineered environments.
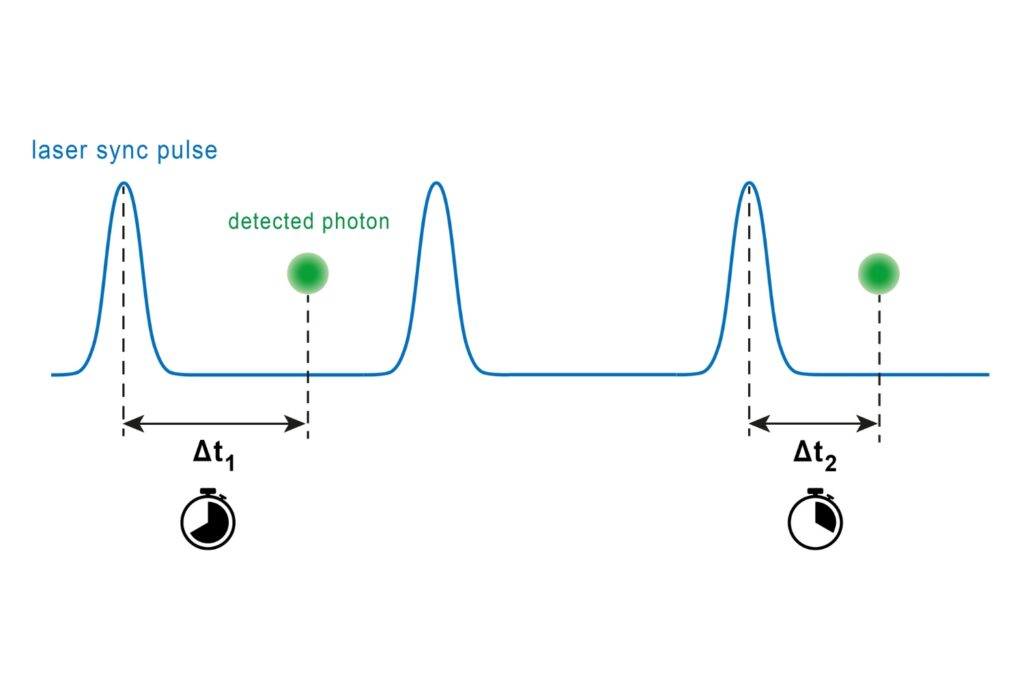
A photon timing technique that measures single-photon arrival times to resolve ultrafast dynamics in fluorescence, materials research, and quantum optics.
Förster Resonance Energy Transfer (FRET) is a fluorescence-based technique that measures non-radiative energy transfer from an excited donor fluorophore to a nearby acceptor fluorophore. FRET occurs when both molecules are typically within 1–10 nm and their emission and absorption spectra overlap. Because the transfer efficiency depends strongly on the donor–acceptor distance, FRET serves as a molecular ruler at the nanometer scale.
In the life sciences, FRET is widely used to investigate molecular interactions and conformational changes in proteins, nucleic acids, and other biomolecular complexes, enabling quantitative, distance-sensitive measurements both in vitro and in living cells.
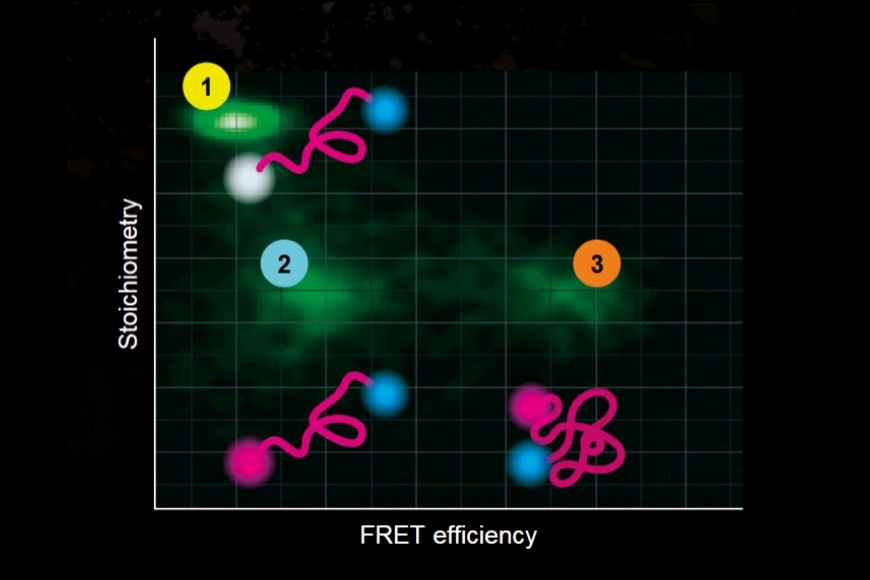
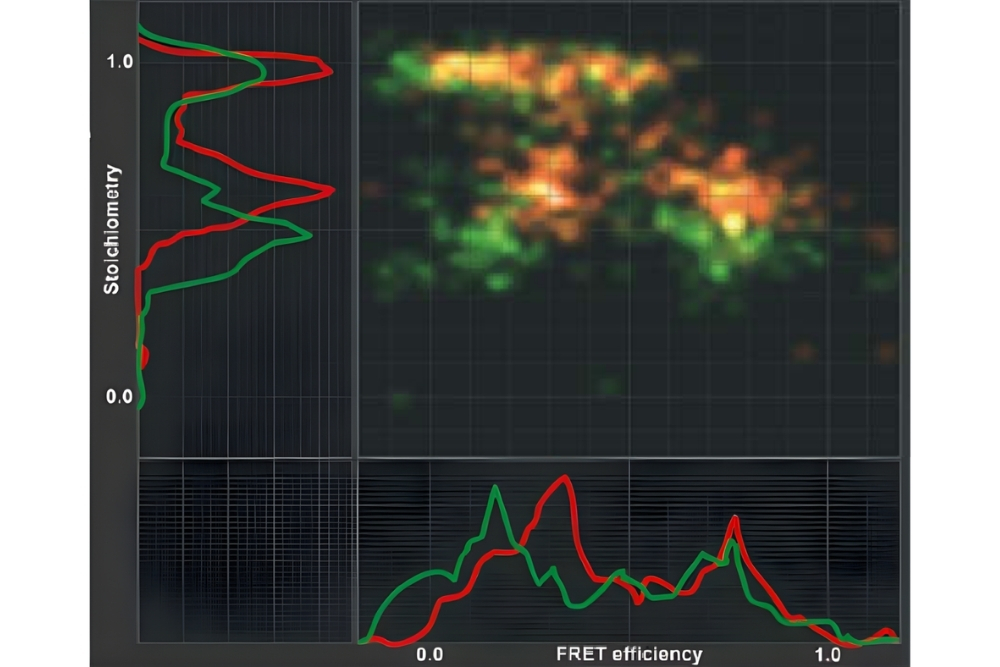
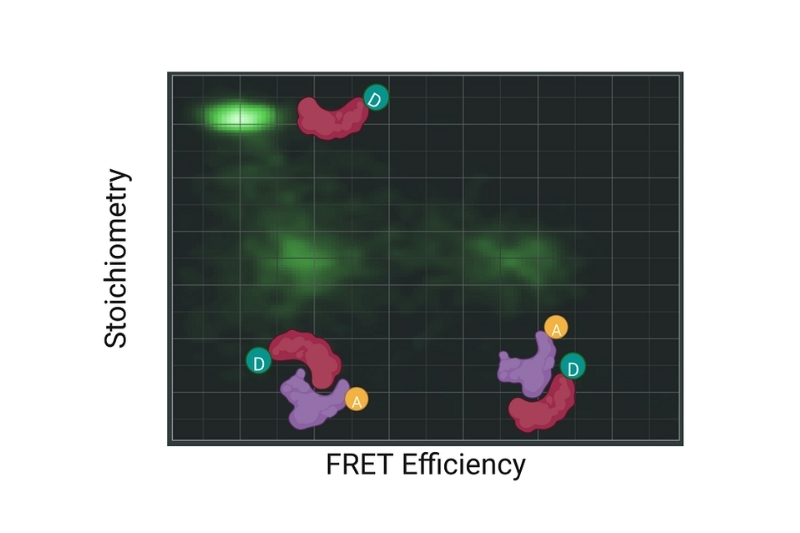 Graphical representation of FRET efficiency plotted against stoichiometry, illustrating different donor and acceptor populations and their relative contributions in a FRET experiment.
Graphical representation of FRET efficiency plotted against stoichiometry, illustrating different donor and acceptor populations and their relative contributions in a FRET experiment.Förster Resonance Energy Transfer (FRET) is a non-radiative energy transfer process between an excited donor fluorophore and a nearby acceptor fluorophore mediated by long-range dipole–dipole coupling. Upon donor excitation, its energy is transferred directly to the acceptor without photon emission, provided both fluorophores are in close proximity and exhibit sufficient spectral overlap.
The efficiency of FRET depends on the donor–acceptor distance, their relative dipole orientation, and the spectral overlap between donor emission and acceptor absorption. Because FRET efficiency decreases steeply with increasing distance, it enables highly sensitive detection of nanometer-scale changes in molecular spacing.
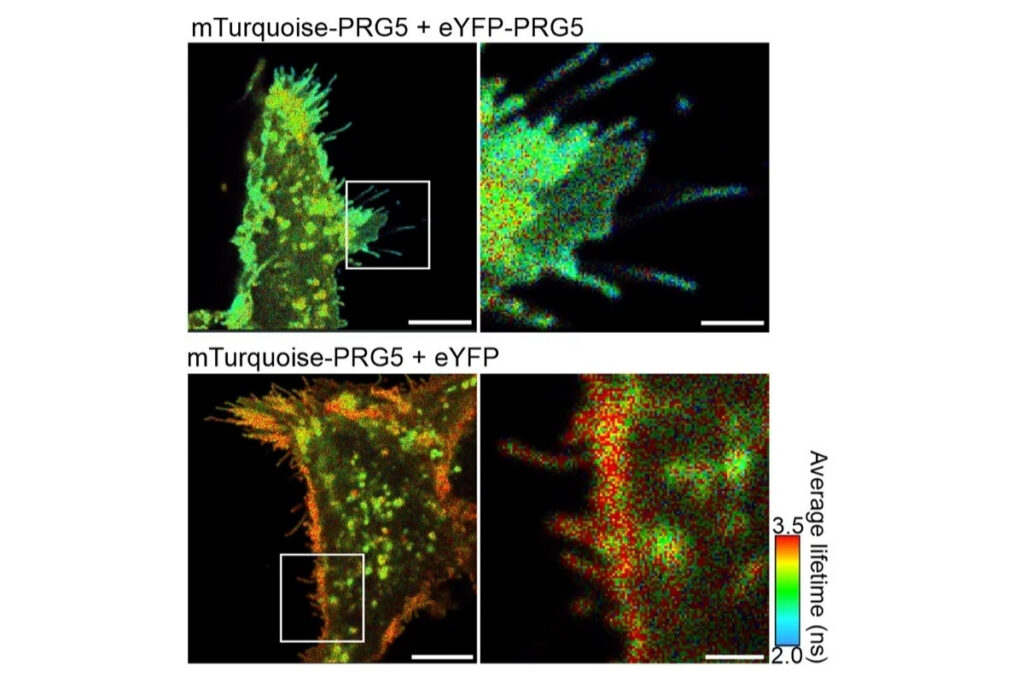
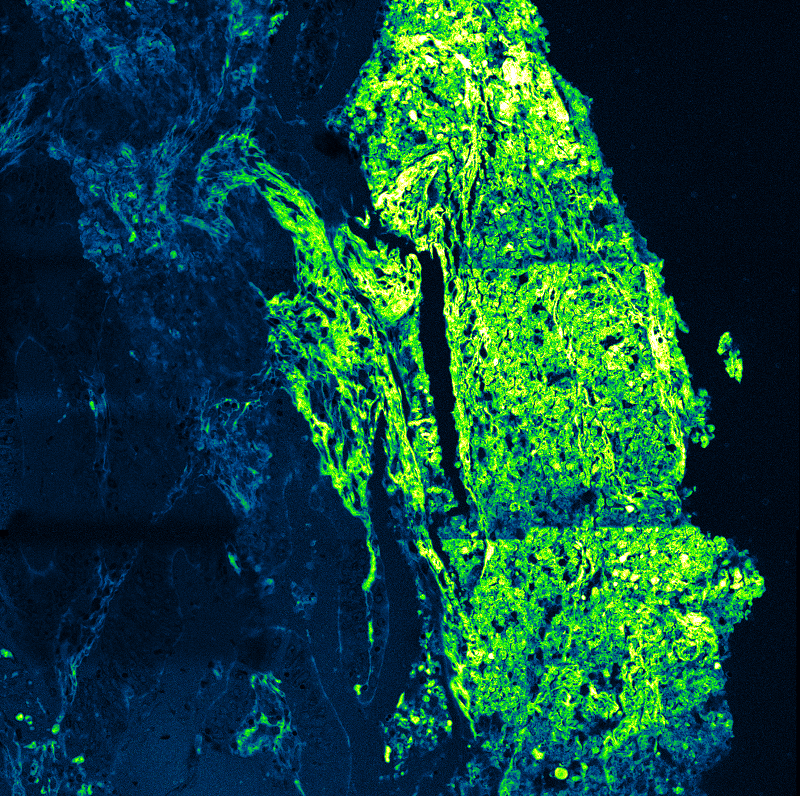
 FLIM-FRET measurements of SNAP33 fused to mVenus co-expressed with different SNARE proteins (SYP21, SYP111, SYP121, SYP122) in living plant cells. Donor fluorescence lifetime values indicate protein-protein interactions through FRET-dependent lifetime reduction.
FLIM-FRET measurements of SNAP33 fused to mVenus co-expressed with different SNARE proteins (SYP21, SYP111, SYP121, SYP122) in living plant cells. Donor fluorescence lifetime values indicate protein-protein interactions through FRET-dependent lifetime reduction.Förster Resonance Energy Transfer (FRET) enables quantitative detection of molecular interactions and conformational changes with nanometer-scale sensitivity. By translating distance changes into measurable variations in FRET efficiency, the method provides direct access to molecular proximity beyond the diffraction limit. FRET can be applied in vitro and in living cells, allowing interaction dynamics and structural rearrangements to be studied under physiologically relevant conditions.
Reliable Förster Resonance Energy Transfer (FRET) measurements require precise optical alignment, sensitive fluorescence detection, and accurate spectral separation of donor and acceptor signals. Because FRET relies on nanometer-scale distance changes, instrument performance directly affects the accuracy of FRET efficiency determination.
Key instrumentation requirements include:
 Luminosa confocal microscope combined with NovaFLIM enables advanced FLIM imaging and analysis workflows.
Luminosa confocal microscope combined with NovaFLIM enables advanced FLIM imaging and analysis workflows.PicoQuant’s microsope systems such as Luminosa combine optimized hardware and integrated software for reliable FRET data acquisition and analysis. Time-resolved platforms such as NovaFLIM and confocal systems like Luminosa provide context-based workflows for FRET experiments, including automated configuration, spectral separation, and quantitative FRET efficiency evaluation. Integrated analysis tools ensure reproducible measurements with proper background correction and cross-talk compensation in vitro and in living cells.
These examples illustrate how FRET enables quantitative analysis of molecular interactions and energy transfer processes in cells and nanoscale systems.
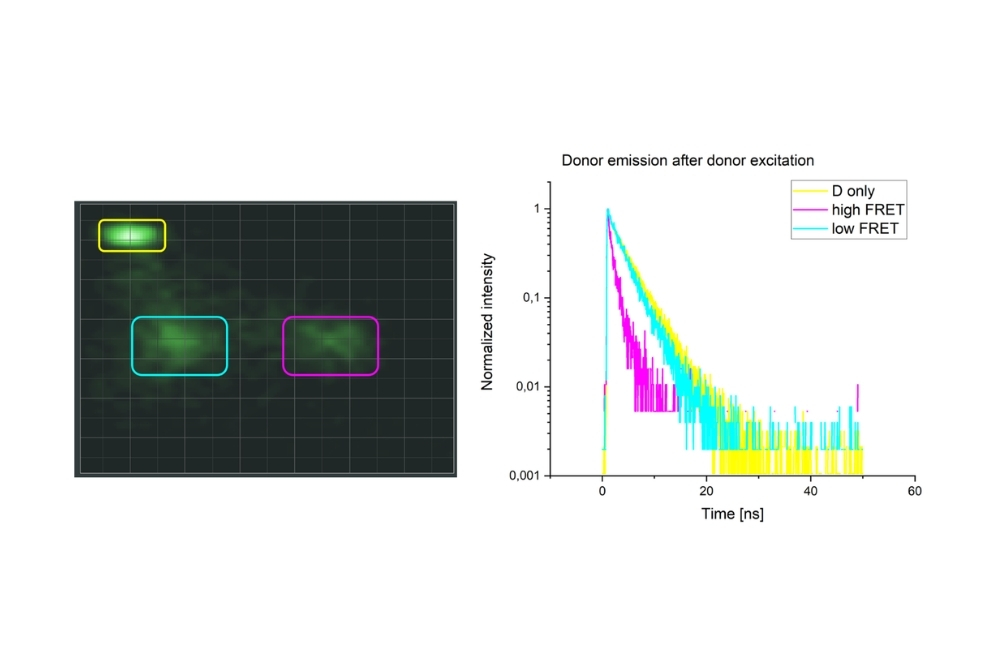
Single-molecule FRET measurements were performed using Luminosa with context-based workflows. Regions of interest selected in raw or corrected E/S histograms enabled burst-resolved fluorescence lifetime analysis across all detection channels, including donor and acceptor emission. Multi-exponential fitting provided quantitative insight into FRET populations and structural heterogeneity.
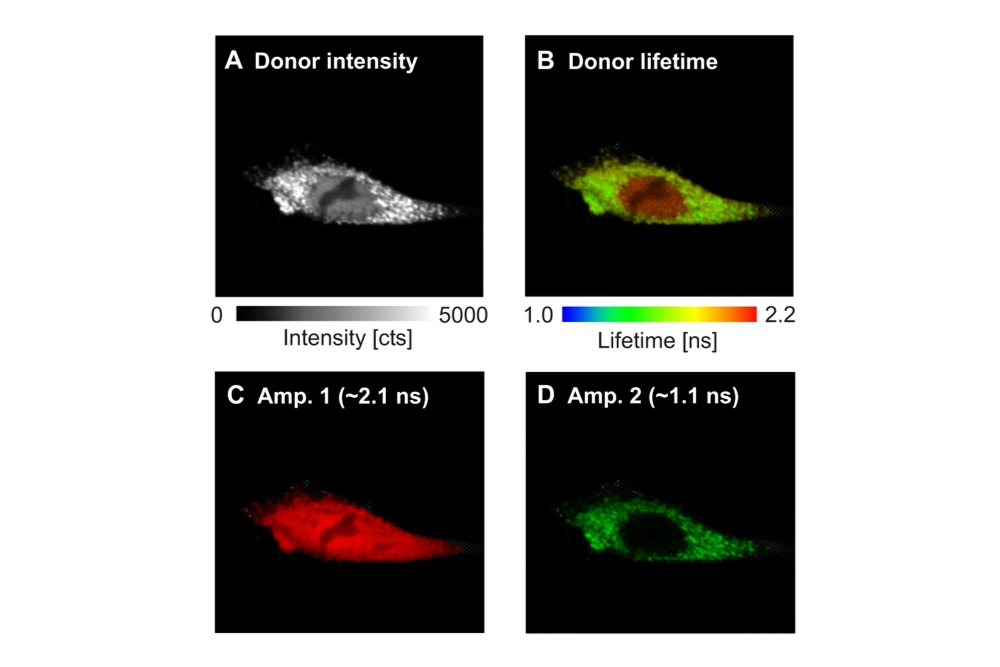
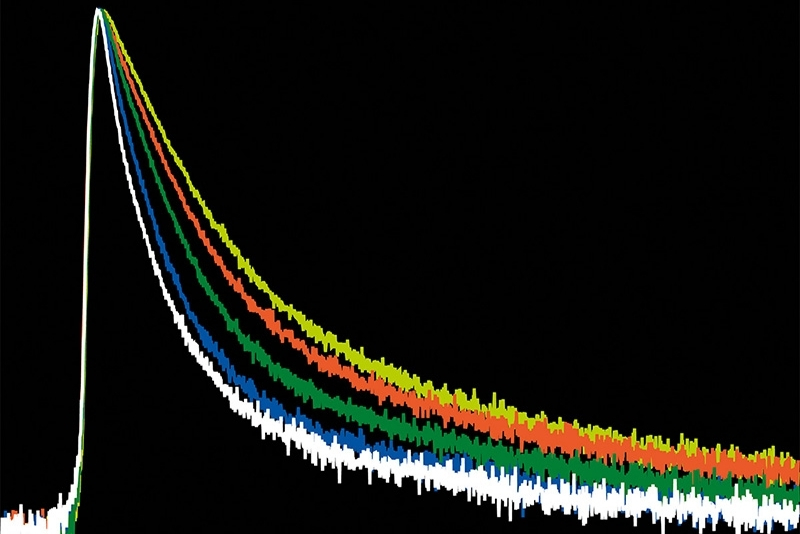
FLIM-FRET was used to analyze the interaction between GFP-N-WASP and RFP-TOCA-1 in cells using an Olympus FluoView FV1000 equipped with PicoQuant’s LSM Upgrade Kit. Donor lifetime analysis identified quenched and unquenched populations, indicating localization-dependent protein binding in cytoplasmic vesicles.
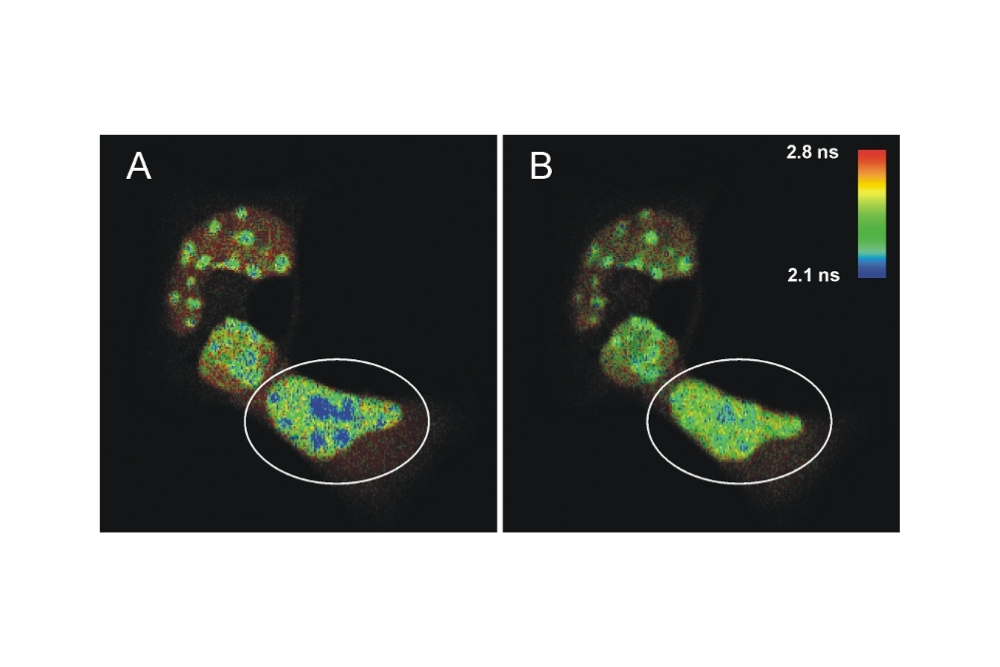
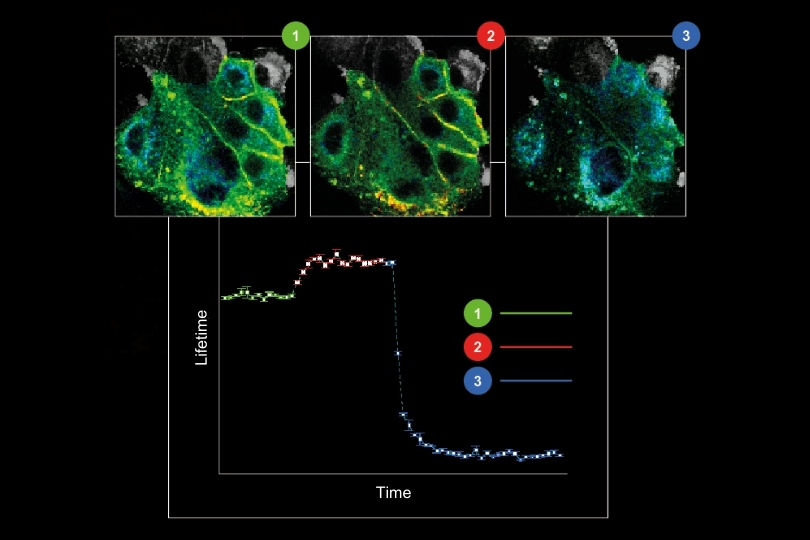
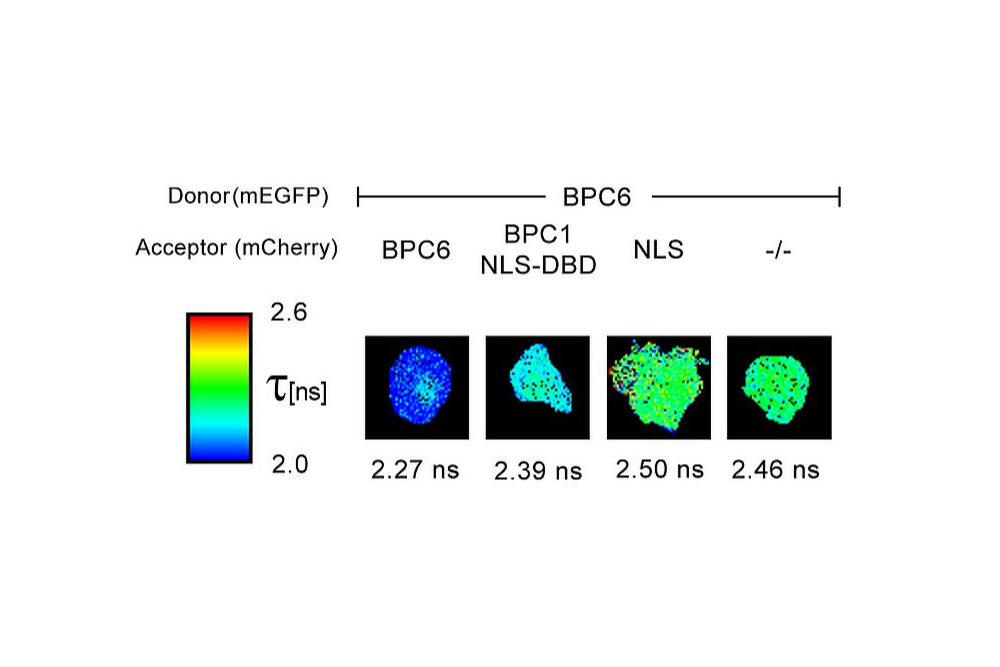
FLIM-FRET measurements were performed on living mouse cells using an Olympus FluoView FV1000 equipped with a PicoQuant LSM Upgrade Kit. Donor lifetime shortening of ECFP-C/EBPα DBD indicated dimer formation at pericentric heterochromatin. Acceptor photobleaching restored donor lifetime, confirming FRET-dependent intra-nuclear protein interaction.
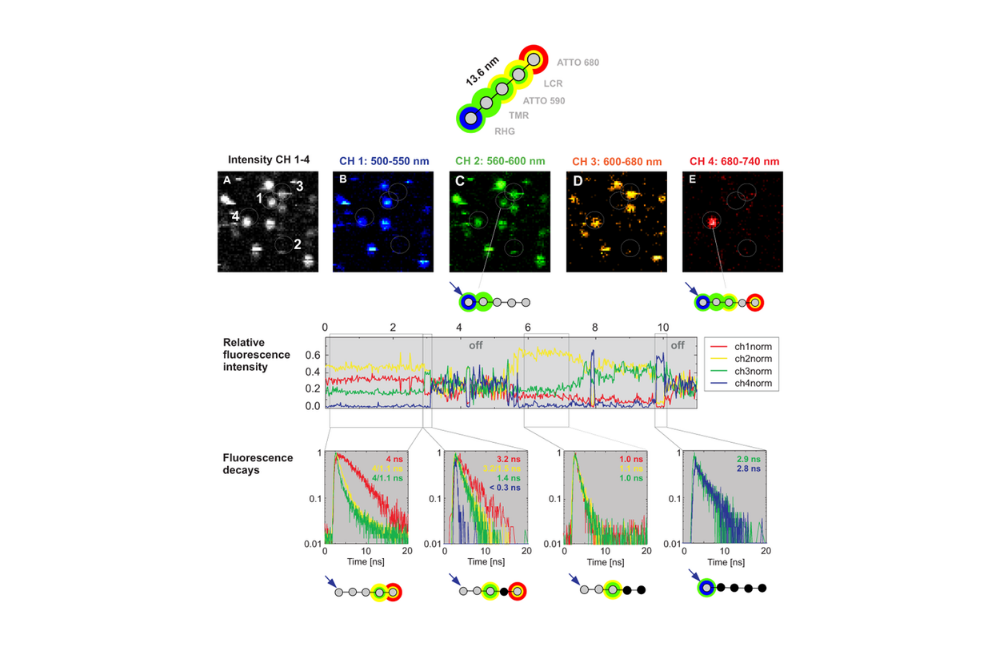
Multistep Förster Resonance Energy Transfer was analyzed in a DNA-based photonic wire labeled with multiple dyes using a MicroTime 200 system. Spectrally resolved detection combined with time-correlated single photon counting enabled stepwise FRET efficiency analysis and fluorescence lifetime characterization of individual emitters within the multichromophoric structure.
Single-Molecule FRET (smFRET) detects FRET events at the level of individual donor-acceptor pairs. By analyzing fluorescence bursts from single donor–acceptor complexes, smFRET reveals structural heterogeneity, dynamic conformational changes, and molecular interactions that are hidden in ensemble-averaged measurements.
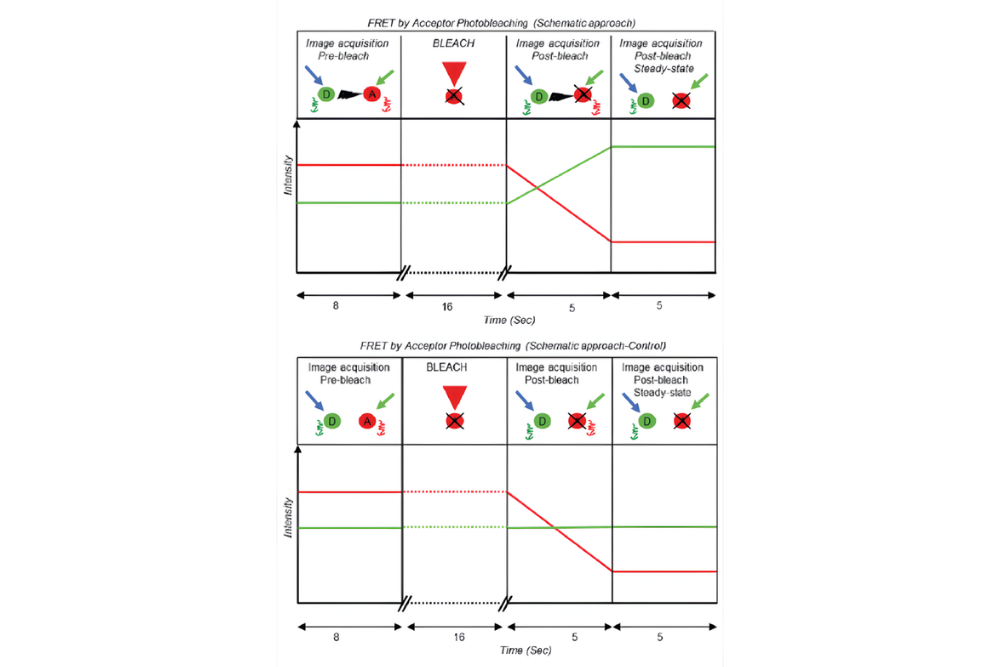
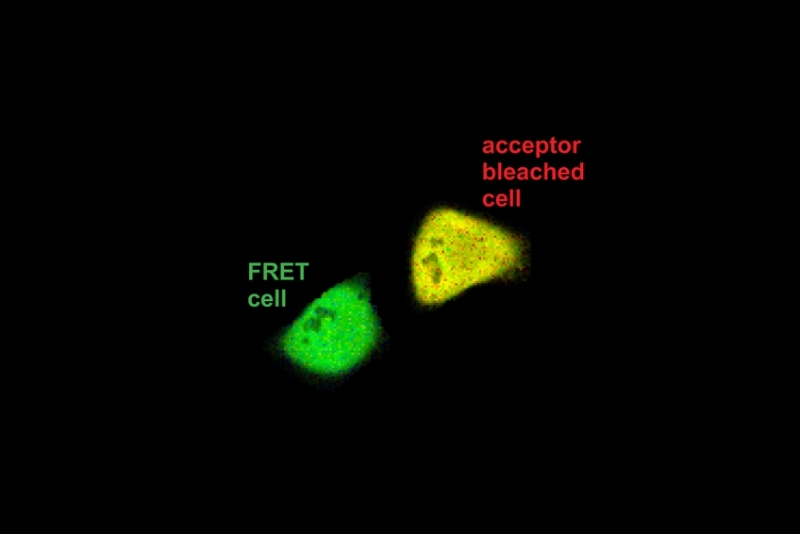
Acceptor Photobleaching FRET (AP-FRET) determines FRET efficiency by selectively photobleaching the acceptor fluorophore and monitoring the resulting increase in donor fluorescence. The loss of energy transfer upon acceptor bleaching directly confirms molecular proximity but is irreversible and limited to endpoint measurements.
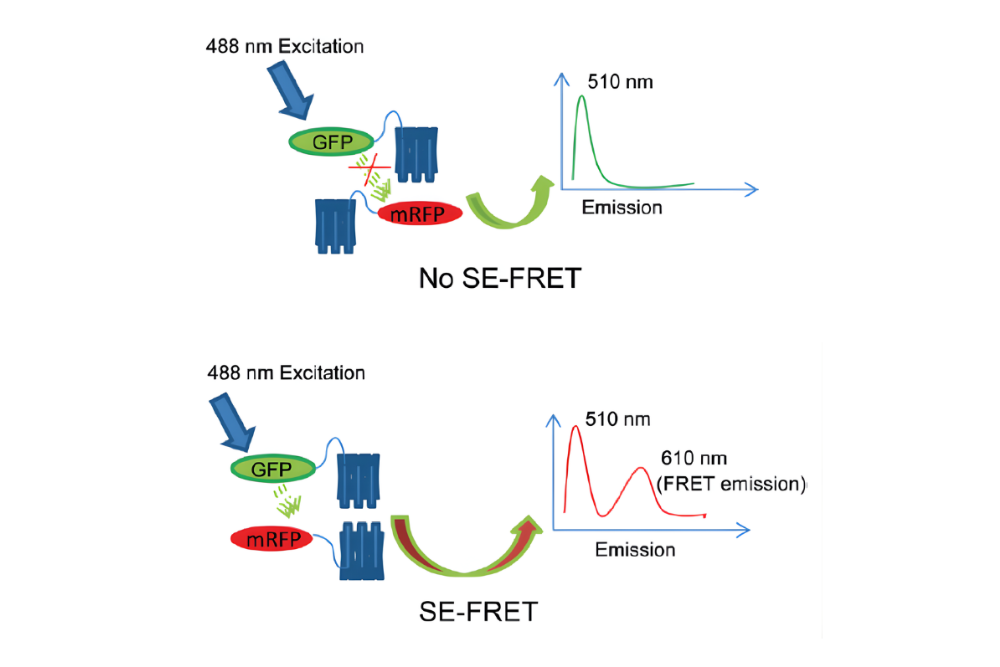
Fluorescence Sensitized Emission FRET (SE-FRET) quantifies FRET by measuring acceptor emission resulting from donor excitation. This intensity-based approach enables spatially resolved FRET imaging but requires careful correction for spectral cross-talk, direct acceptor excitation, and background fluorescence.