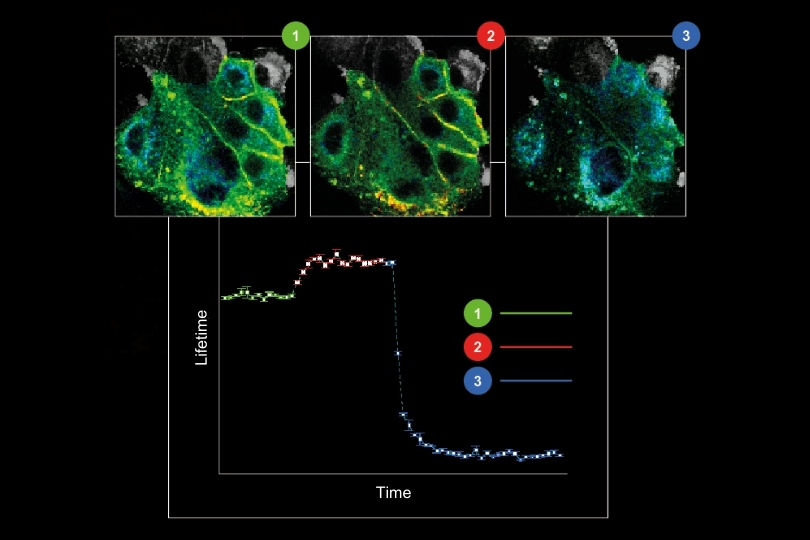
| 09.00 - 09.35 | Chiara Stringari, Palaiseau Cedex, France (Invited Talk) Label-free Fluorescence lifetime microscopy for metabolic imaging across scalesLabel-free Fluorescence lifetime microscopy for metabolic imaging across scales Chiara Stringari Laboratory for Optics and Biosciences (LOB), CNRS, INSERM, École polytechnique, IP Paris, 91128 Palaiseau, France
Fluorescence lifetime imaging microscopy (FLIM) is emerging as a transformative tool in biophotonics. It enables the high-resolution, non-invasive imaging of metabolic processes in live tissues by measuring the lifetime of endogenous biomarkers (such as NAD(P)H and FAD) that are naturally present in cells and tissues, providing a quantitative readout of metabolic pathways, such as glycolysis and oxidative phosphorylation [1, 2]. However, traditional FLIM approaches used to measure and quantify metabolism are limited by factors such as slow acquisition speed, poor signal-to-noise ratio (SNR) and high phototoxicity [3, 4]. In our laboratory, we aim to develop nonlinear optics and FLIM techniques that can measure metabolic transitions at the cellular level across spatial and temporal scales with enhanced speed and performance. In this talk, we will highlight recent advances and applications in label-free metabolic imaging using two-photon fluorescence lifetime microscopy (2P-FLIM) of intrinsic metabolic coenzymes. We discuss here how we implement simultaneous two-photon excitation of NAD(P)H and FAD through wavelength mixing in order to acquire FLIM data of the two biomarkers simultaneously and perform multiparametric metabolic imaging in dynamic biological systems, such as during embryo development and in living tissues [5] and we demonstrated that the two endogenous fluorophores are complementary biomarkers [6]. To improve the interpretation of the FLIM data for metabolic imaging we also developed FLUTE, a Python Graphical User Interface [7]. We then demonstrate how FLIM of metabolic coenzymes reveals the complexity of metabolic processes in intact tissues with minimal phototoxicity and we discuss the optimization and the implementation of this methodology for longitudinal in vivo studies. Finally, we present applications for metabolic imaging in neuroscience, developmental biology, and immunometabolism, measuring subcellular metabolic compartmentalization and complex temporal metabolic patters during stem cell differentiation [8], T cell activation [9] and embryo development. 1. Georgakoudi, I. and K.P. Quinn, Label-Free Optical Metabolic Imaging in Cells and Tissues. Annu Rev Biomed Eng, 2023.
2. Datta, R., et al., Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J Biomed Opt, 2020. 25(7): p. 1-43.
3. Poudel, C., I. Mela, and C.F. Kaminski, High-throughput, multi-parametric, and correlative fluorescence lifetime imaging. Methods Appl Fluoresc, 2020. 8(2): p. 024005.
4. Park, J. and L. Gao, Advancements in fluorescence lifetime imaging microscopy Instrumentation: Towards high speed and 3D. Curr Opin Solid State Mater Sci, 2024. 30.
5. Stringari, C., et al., Multicolor two-photon imaging of endogenous fluorophores in living tissues by wavelength mixing. Sci Rep, 2017. 7(1): p. 3792.
6. Ung, T.P.L., et al., Simultaneous NAD(P)H and FAD fluorescence lifetime microscopy of long UVA-induced metabolic stress in reconstructed human skin. Sci Rep, 2021. 11(1): p. 22171.
7. Gottlieb, D., et al., FLUTE: a Python GUI for interactive phasor analysis of FLIM data Biological Imaging., 2023. 3:e21.
8. Sánchez-Ramírez, E., et al., Coordinated metabolic transitions and gene expression by NAD+ during adipogenesis. J Cell Biol., 2022. 221(12).
9. Paillon, N., et al., Label-free single-cell live imaging reveals fast metabolic switch in T lymphocytes. 2024. 35(1): p. ar11. |
| 09.35 - 09.55 | Fabian Ruebsamen, Bochum, Germany (Student Award) Assessing the membrane properties of Oli-neu cells using FLIM and labelled lipidsAssessing the membrane properties of Oli-neu cells using FLIM and labelled lipids Fabian Ruebsamen1, Annika Haak2, Thomas Günther-Pomorski1 1Department of Biochemistry II, Ruhr-Universität Bochum, Bochum, Germany
2RG Nanoscopy, RUBION, Ruhr-Universität Bochum, Bochum, Germany
Oligodendrocytes are the myelinating cells of the central nervous system, which insulate neurons. During the first three postnatal weeks, oligodendrocytes undergo a tightly regulated differentiation process from oligodendrocyte precursor cells into mature, myelin-producing cells. This transition is accompanied by drastic changes in plasma membrane composition and membrane properties required to enable the formation of the highly specialised myelin-containing membranes. In this study, the secondary mouse oligodendroglial cell line Oli-neu was employed as an in-vitro model to investigate membrane dynamics during oligodendrocyte maturation: Membrane viscosity and lipid behaviour were examined using fluorescence lifetime imaging microscopy (FLIM)-based viscosity probes and fluorescently labelled lipid derivatives. These complementary techniques enabled the characterisation of membrane microenvironment properties and lipid movement. |
| 09.55 - 10.15 | Noah H. Salama, Düsseldorf, Germany (Student Award) Mapping Structurally Variable DNA Constructs with FRET Nanoscopy and Optical PythagorasMapping Structurally Variable DNA Constructs with FRET Nanoscopy and Optical Pythagoras Noah H. Salama1, Nicolaas T. M. van der Voort1, Jan-Hendrik Budde1, Michelle P. Rademacher2, Suren Felekyan1, Christian A. Hanke1, Alexander Henrichs1, Ralf Kühnemuth1, Anders Barth3, Claus A. M. Seidel1 1Chair for Molecular Physical Chemistry, Heinrich-Heine-University Düsseldorf, Germany
2Institute for Physical Chemistry II, Heinrich-Heine-Universität Düsseldorf, Germany
3Department of Bionanoscience, Delft University of Technology, Delft, Netherlands
Recently, the lateral resolution of super-resolution microscopy reached the size of individual molecules.[1] Nevertheless, obtaining precise information on 3D orientation of structures remains challenging.
We established an approach combining colocalization stimulated emission depletion (cSTED) microscopy with concurrent analysis of Förster resonance energy transfer (FRET) to identify heterogeneities in DNA systems.[2] We obtained precise lateral and axial distances using the Optical Pythagoras by comparing the localization distance between two fluorophores from cSTED to the Euclidean distance based on a structure model or obtained from FRET analysis. We simultaneously localize both fluorophores of single FRET pairs, yielding sample orientation in 3D, with sub-nanometer localization precision for individual dyes. Scanning 1x1 µm in a few seconds, we reached an average distance precision of 1.4 nm for a donor and acceptor label coupled to a single molecule. Studying immobilized dsDNA rulers, bound to the surface from one or both ends, we measure a mean inclination angle of 64° and 34°, respectively. The latter angle correlates well with the local roughness of the surface found by AFM measurements.
We apply our workflow on the level of single particles, enabling us to map conformational exchange within the 3D structure of DNA Holliday junctions. [1] Hell, S. W. et al., Science, 355, 606-612 (2017).
[2] Budde, J.-H. et al., arXiv preprint, doi.org/10.48550/arXiv.2108.00024 (2022). |
| 10.15 - 10.35 | |
| 10.35 - 10.45 | |
| 10.45 - 11.20 | |
| |
| 11.20 - 11.50 | Johannes Stein, Berlin, Germany (Invited Talk) Integrating DNA-PAINT with electron microscopy for ultrastructural molecular imagingIntegrating DNA-PAINT with electron microscopy for ultrastructural molecular imaging Johannes Stein Emmy Noether Group Leader Max Planck Institute for Molecular Genetics
DNA-PAINT enables highly multiplexed molecular imaging in cells, yet the resulting maps remain largely disconnected from cellular ultrastructure. Electron microscopy resolves cellular architecture at nanometer resolution but lacks molecular specificity. Bridging these views has been a longstanding challenge.
We present a correlative workflow combining kinetically enhanced DNA-PAINT (tkPAINT) with transmission electron microscopy (TEM). Cellular ultrastructure is preserved during cryosectioning, labeling, and multiplexed Exchange-PAINT imaging, followed by TEM processing of the same sections. This allows nanometer-scale registration of single-protein localizations directly onto defined ultrastructural features.
Biomolecules can thus be assigned to specific membranes and organelle subdomains within the same specimen, enabling molecular organization to be interpreted in their structural context. Our approach links highly multiplexed single-molecule imaging with electron microscopy and provides a promising route toward ultrastructure-resolved cell biology. |
| 11.50 - 12.10 | Nicco Corduri, Fribourg, Switzerland (Student Award) DNA-PAINT super-resolution microscopy for carbon nanotube identificationDNA-PAINT super-resolution microscopy for carbon nanotube identification Nicco Corduri1, Alan Szalai2, Karol Kołątaj1, Guillermo Acuna1 1University of Fribourg
2Centro de Investigaciones en Bionanociencias, CIBION / University of Fribourg
Super-resolution microscopy has become indispensable for investigating nanoscale architectures beyond the diffraction limit of conventional optical systems, ultimately reaching 1-10 nm spatial resolution. However, fluorescent based super-resolution imaging of carbon nanotube (CNT)-based assemblies has mostly relied on inducing blinking on the CNT’s NIR photoluminescence, with a resolution limited to several tens of nanometers [1],[2]. Here, we present a DNA-PAINT [3],[4] imaging strategy developed for DNA-functionalized CNTs and apply it to the quantitative characterization of both isolated nanotubes and complex assemblies organized on DNA origami platforms. Remarkably, DNA-PAINT offers nanometer-scale localization precision combined with multiplexing capabilities. We first validate our method on isolated CNTs, reliably super-resolving individual nanotubes. The achieved localization precision of 2- 3 nm allows direct detection of individual docking sites along the CNTs. Notably, we observe a direct dependence of the spacing between docking sites on the length of the functionalizing DNA strands. Building on these results, we investigate DNA origami-directed assemblies [5],[6], where large rectangular templates position multiple CNT populations carrying different DNA sequences. Sequential DNA-PAINT imaging allows selective visualization of each CNT population. This approach allows the accurate reconstruction of two-dimensional arrays containing up to nine parallel CNTs with center-to-center separations as small as 14 nm. [1] Lambert, B. P., Kerkhof, H., Flavel, B. S. & Cognet, L. ACS Nano 18, 30728–30736 (2024).
[2] Otsuka, K., Ishii, A. & Kato, Y. K. Opt. Express 27, 17463–17473 (2019).
[3] Schnitzbauer, J., Strauss, M. T., Schlichthaerle, T., Schueder, F. & Jungmann, R. Nat. Protoc. 12, 1198–1228 (2017).
[4] Jungmann, R. et al. Nano Lett. 10, 4756–4761 (2010).
[5] Rothemund, P. W. K. Nature 440, 297–302 (2006).
[6] Maune, H. T. et al. Nat. Nanotechnol. 5, 61–66 (2010). |
| 12.10 - 12.30 | Bela T. L. Vogler, Jena, Germany (Student Award) MINFLUX-enabled 3D single-particle tracking reveals live-cell neuronal membrane topographyMINFLUX-enabled 3D single-particle tracking reveals live-cell neuronal membrane topography Bela T. L. Vogler1,2, Christian Eggeling1,2,3 1Institute of Applied Optics and Biophysics, Friedrich-Schiller University Jena, Jena, Germany.
2Department of Biophysical Imaging, Leibniz Institute of Photonic Technologies e.V., Jena, Germany, Member of the Leibniz Centre for Photonics in Infection Research (LPI), Jena, Germany
3Jena Center for Soft Matter, Jena, Germany.
MINFLUX fluorescence microscopy enables nanometer-precision localization at microsecond temporal resolution, offering unique opportunities for three-dimensional (3D) single-particle tracking (SPT). Here, we implement 3D MINFLUX tracking in live neurites to resolve the motion of quantum dot–labelled GPI-GFP molecules on curved tubular membranes. Kilohertz sampling permits analysis of membrane dynamics across timescales from microseconds to seconds. The true 3D tracking capability allows us to quantify diffusion along the tubular axis and to detect periodic confinement with a characteristic spacing of ~200 nm. This spacing is consistent with diffusion barriers imposed by the submembrane actin–spectrin lattice. Our results demonstrate that 3D MINFLUX SPT provides robust and quantitative access to nanoscale membrane organization in complex cellular geometries. |
| 12.30 - 12.50 | Adrian Platz, Jena, Germany (Student Award) A photometric pinhole: event-based optical sectioning without demodulationA photometric pinhole: event-based optical sectioning without demodulation Adrian Platz, Gabriel Baum, Andreas Stark, Christian Franke Institute of Applied Optics and Biophysics, Friedrich-Schiller Universität Jena
Optical sectioning in widefield fluorescence microscopy currently relies on physical apertures or computational demodulation of structured illumination. Demodulation methods fail catastrophically in scattering tissue when the pattern model breaks down in 3D cell cultures, tissue sections, and organoids.
Here, we introduce the event-based photometric pinhole. Therefore, homogeneous illumination with distinct intensity minima (holes) is projected onto the sample, and fluorescence is detected with an event-based camera. The camera's hardware log-contrast threshold θ acts as an axial gate: in focus, the hole produces contrast exceeding θ and triggers events; out of focus, diffraction reduces contrast below θ. The gate is pattern-agnostic: each pixel evaluates its local contrast independently. Scattering can blur a hole but cannot invert it: a hole stays a hole, and sectioning degrades gracefully with depth rather than producing artefacts.
At dhole = 3.5 µm we achieve ~600 nm optical sectioning at 640 nm illumination, comparable to Airyscan- and confocal microscopy. Furthermore, we present a forward model that predicts sub-250 nm optical sectioning for an optimized illumination system.
We demonstrate sectioning in fixed cell cultures, thick brain sections, and glioblastoma tumoroids. The method requires no scanning and no pattern knowledge. |
| 12.50 - 13.10 | Alexandra Kaminer, Frankfurt, Germany (Student Award) Quantitative mapping of nanoscale protein assemblies by DNA-PAINTQuantitative mapping of nanoscale protein assemblies by DNA-PAINT Alexandra Kaminer, Yunqing Li, Jasmin Janzen, Marina Dietz, Mike Heilemann
Activation of receptor tyrosine kinases begins with ligand-induced receptor engagement at the cell surface, leading to the formation of membrane-associated signaling assemblies that activate intracellular signal transduction[1]. Determining the molecular composition and spatial organization of these assemblies in situ remains challenging due to their nanoscale dimensions and intrinsic heterogeneity. Here, we present a single-molecule super-resolution imaging (DNA-PAINT) and analysis workflow that enables the identification and quantitative characterization of individual signaling assembly sites in cells. Using this approach, we examine the nanoscale reorganization of the epidermal growth factor receptor (EGFR) together with its adaptor protein Grb2 following stimulation with the physiological ligand EGF[2]. Furthermore, we apply this workflow to determine the molecular architecture of MET-receptor assemblies at the plasma membrane in a ligand-stimulation dependent manner[3], [4]. [1] Lemmon, M. A. & Schlessinger, J. Cell 141, 1117–1134 (2010).
[2] Kaminer, A., Li, Y., Barth, H.-D., Dietz, M. S. & Heilemann, M. ChemPhysChem 27, e202500915 (2026).
[3] Niemann, H. H. European Journal of Cell Biology 90, 972–981 (2011).
[4] Li, Y., Arghittu, S.M., Dietz, M.S. et al. Nat Commun 15, 9486 (2024). |
| 13.10 - 14.40 | |
| Biological Applications 2 |
| 14.40 - 15.10 | Michelle S. Frei, Zürich, Switzerland (Invited Talk) Chemigenetic tools for live-cell fluorescence microscopyChemigenetic tools for live-cell fluorescence microscopy Michelle S. Frei Institut für Chemie und Angewandte Biowissenschaften ETH Zürich
Synthetic fluorophores, in combination with self-labeling protein tags, have become indispensable tools in fluorescence microscopy. Compared to fluorescent proteins, synthetic fluorophores show enhanced brightness, photostability, and are available in the far-red to near-infrared spectral region, which is beneficial for advanced fluorescence microscopy applications including super-resolution microscopy. Moreover, fluorogenic fluorophores, which become fluorescent only upon target binding, are particularly well suited for live-cell applications due to their high signal-to-background ratios. While synthetic chemistry allows fine-tuning of a fluorophore’s photophysical properties, the protein environment surrounding the fluorophore binding site also plays a crucial role. Here, we demonstrate how protein engineering of the self-labeling protein HaloTag can be harnessed to develop innovative tools for live-cell fluorescence microscopy.
First, we present a series of engineered HaloTag variants with altered brightness and fluorescence lifetime characteristics that enable multiplexed imaging of up to three distinct targets with in a single spectral channel using fluorescence lifetime imaging microscopy (FLIM). Second, we explore the use of HaloTag beyond its conventional role as a labeling tag by integrating it into functional biosensors. Specifically, we sandwiched a circularly permuted version of HaloTag7 between a phosphoamino acid binding protein and a protein kinase A (PKA) specific peptide to get access to HaloTag-based kinases activity reporters (HaloKARs). Through protein engineering, we optimized the biosensor’s performance ultimately reaching a 12-fold change upon PKA stimulation. The best HaloKAR was successfully applied in advanced multiplexing experiments and functional super-resolution microscopy measurements.
Overall, our work underscores the power of combining protein engineering with synthetic fluorophores to create imaging tools with outstanding properties. We expect these HaloTag-based tools to facilitate the widespread use of advanced microscopy techniques. |
| 15.10 - 15.30 | Daria Maksutova, Dortmund, Germany (Student Award) Towards quantitative predictions of lifetime and anisotropy values of fluorophores on biomoleculesTowards quantitative predictions of lifetime and anisotropy values of fluorophores on biomolecules Daria Maksutova, Thomas-Otavio Peulen, Thorben Cordes Biophysical Chemistry, Department of Chemistry and Chemical Biology, Technische Universität Dortmund, Germany
Fluorescent labelling enables powerful fluorescence-based approaches, including single-molecule detection and Förster resonance energy transfer (FRET), to investigate biomolecular structure and function [1,2]. Quantitative interpretation of such experiments depends on the photophysical properties of attached dyes, which are modulated by local biomolecular and solvent environments and remain difficult to assess and predict [3]. Our work aims to establish a predictive framework for fluorescence parameters of dyes on biomolecules, accounting for local environmental effects.
To disentangle dye–protein and dye–solvent interactions, we investigate dyes from different structural classes attached to E. coli maltose-binding protein, a well-characterized test system. In multiparametric experiments down to the single-molecule level, we combine time-resolved and anisotropy-resolved fluorescence with fluorescence correlation spectroscopy to probe quenching processes from the pico- to millisecond range [4]. Solvent-mediated quenching contributions [5] are assessed simultaneously for red and far-red dyes by comparing measurements in aqueous and deuterated buffers.
By correlating fluorophore and protein structural features with experimental fluorescence parameters [3], we establish the basis for a coarse-grained dye model that captures solvation- and amino-acid-mediated quenching via photoinduced electron transfer [4]. Our results reveal pronounced site-dependent variations in fluorescence lifetime and anisotropy, underscoring the need for predictive models to guide fluorescence experiment design. [1] G. Agam et al., Nature Methods, 20.4, 523–535 (2023).
[2] C. Gebhardt et al., Nature Communications, 16.1, 4147 (2025).
[3] T.-O. Peulen, D. Maksutova, and T. Cordes, arXiv:2602.21931 (2026).
[4] T.-O. Peulen, O. Opanasyuk, and C. A. M. Seidel, The Journal of Physical Chemistry B, 121.35, 8211–8241 (2017).
[5] J. Maillard et al., Chemical Science, 12.4, 1352–1362 (2021). |
| 15.30 - 15.50 | Scott C. Blanchard, MEMPHIS, United States Parallel stopped-flow interrogation of diverse biological systems at the single-molecule scaleParallel stopped-flow interrogation of diverse biological systems at the single-molecule scale
Scott C. Blanchard*1,2,
Roman Kiselev+1,
Ryan A. Brady+1,
Arnab Modak1,
Jose L. Alejo1,
Aaron Cruz-Navarrete1,
Daniel S. Terry1,
Roger B. Altman1,
Wesley B. Asher3,4,
Jonathan A. Javitch3,4,5
1Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN, USA
2Department of Chemical Biology & Therapeutics, St. Jude Children’s Research Hospital, Memphis, TN, USA
3Department of Psychiatry, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, USA
4Division of Molecular Therapeutics, New York State Psychiatric Institute, New York, NY, USA
5Department of Molecular Pharmacology and Therapeutics, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, USA
*Corresponding Author
+These authors contributed equally
Single-molecule imaging techniques have provided unprecedented insights into functional changes in composition and conformation across diverse biological systems. As with other biophysical methods, single-molecule fluorescence and Förster resonance energy transfer investigations are typically limited to examination of one sample at a time. Consequently, experimental throughput is restricted, and experimental variances are introduced that can obscure functional distinctions in closely related systems. Here, to address these limitations, we introduce parallel rapid exchange single-molecule fluorescence and single-molecule Förster resonance energy transfer to enable simultaneous steady-state and pre-steady-state interrogations of diverse systems. Using this approach, we elucidate the timing of distinct conformational events underpinning β-arrestin1 activation, unmask antibiotic-induced impacts on messenger RNA decoding fidelity and demonstrate that endogenously encoded ribosomal RNA sequence variation modulates antibiotic sensitivity. This generalizable and scalable method promises to broaden the scope and reproducibility of quantitative single-molecule interrogations of biomolecular function. Kiselev R, Brady RA, Modak A, Cruz-Navarrete FA, Alejo JL, Terry DS, Altman RB, Asher WB, Javitch JA, Blanchard SC. Parallel stopped-flow interrogation of diverse biological systems at the single-molecule scale. Nat Methods. 2026 Jan;23(1):78-87. doi: 10.1038/s41592-025-02944-4. Epub 2025 Dec 2. PMID: 41331139; PMCID: PMC12791014. |
| 15.50 - 16.10 | Eike Wienbeuker, Osnabrück, Germany (Student Award) Metal-induced energy transfer uncovers activation-induced axial reorganization of signaling complexes inside cellsMetal-induced energy transfer uncovers activation-induced axial reorganization of signaling complexes inside cells Eike Wienbeuker1, Arthur Felker1, Oleksii Nevskyi2, Steffen T. Harms1, Daniel Marx2, Kevin Tanzusch1, Anna Chizhik2, Rainer Kurre1, Changjiang You1, Jörg Enderlein2, Jacob Piehler1 1Department of Biology/Chemistry and Center for Cellular Nanoanalytics, Osnabrück University, Barbarastr. 11, Germany
2Third Institute of Physics (Biophysics), Georg August University, Friedrich-Hund-Platz 1, 37077 Göttingen, Germany
Transmembrane signaling mediated by cytokine receptors orchestrates key cellular processes such as proliferation, differentiation, and immune responses. While numerous high-resolution structures of cytokine receptor ectodomains are available, the structural organization of the largely disordered intracellular domains (ICD) has remained unclear. Here, we interrogate the axial organization of cytokine receptor signaling complexes at the plasma membrane by metal-induced energy transfer (MIET). For this purpose, we leveraged biofunctionalized nanodot arrays (bNDAs) to capture cell surface receptors at a defined distance from the substrate. This approach enables axial distance measurements with ~1 nm resolution at both ensemble and single-molecule levels via fluorescence lifetime imaging microscopy (FLIM). Using the prototypic class I cytokine receptor GP130 as a model system, we uncover by MIET that the ICD extends randomly into the cytosol in the resting state but undergoes an axial rearrangement upon signal activation. These results highlight the potential of bNDA-supported MIET for resolving the axial architecture of signaling complexes within the cellular context. |
| 16.10 - 16.45 | |
| Sensors & Material Science Applications |
| 16.45 - 17.05 | Kristina Djordjevic, Munich, Germany (Student Award) DNA Origami-Based Plasmonic Nanoantennas for Amplification-Free Single-Molecule microRNA DetectionDNA Origami-Based Plasmonic Nanoantennas for Amplification-Free Single-Molecule microRNA Detection Kristina Djordjevic, Julian Bauer, Renukka Yaadav, Philip Tinnefeld Department of Chemistry and Center for NanoScience, Ludwig-Maximilians- University, Butenandtstraße 5−13, 81377, Munich, Germany
DNA origami-based plasmonic nanoantennas have emerged as a powerful platform for amplification-free single-molecule fluorescence detection by combining programmable DNA nanotechnology with the electromagnetic field enhancement of metallic nanoparticles.1 Here, we present DNA origami nanoantenna architectures for ultrasensitive nucleic acid detection, with a particular focus on microRNA (miRNA) sensing for future point-of-care diagnostics. The structures are based on DNA origami “Fork” geometries that enable nanometer-precise positioning of silver nanoparticles and nucleic acid assay components within plasmonic hotspots.2,3 Upon target hybridization, fluorescent imaging strands localize in the hotspot region, resulting in strongly enhanced fluorescence signals detectable at the single-molecule level by confocal and TIRF microscopy. Different assay configurations were systematically investigated to evaluate sensitivity, specificity, kinetics, and structural stability. In addition to proof-of-concept nucleic acid detection, this work explores amplification-free miRNA sensing motivated by the diagnostic relevance of short regulatory RNAs in diseases such as cancer and endometriosis. Future studies will focus on systematic mismatch investigations to evaluate sequence discrimination and single-base mismatch sensitivity under plasmon-enhanced conditions. Furthermore, nanopatterning strategies will be expanded toward multiplexed nanoantenna arrays for high-density single-molecule biosensing.3,4 [1] Viktorija Glembockyte, Lennart Grabenhorst, Kateryna Trofymchuk et al., Acc. Chem. Res. 54, 3338-3348 (2021).
[2] Cindy Close et al., Adv. Mater. Interfaces, 9, 2200255 (2022).
[3] Renukka Yaadav et al., Adv. Mater., 37, 07407 (2025).
[4] Shetty R. M. et al., J. ACS Nano, 15, 11441-11450 (2021).
|
| 17.05 - 17.25 | Eva Münzel, München, Germany (Student Award) Tackling the Speed Limit of Brownian Computation |
| 17.25 - 17.45 | Maarten H. van der Hoeven, Berlin, Germany (Student Award) Widefield Fluorescence Microscopy for Quantum Materials Science |
| 17.45 - 17.55 | |
| 18.20 - | |