

Key Highlights
- Super-resolution FRET imaging is achieved by combining single-molecule localization microscopy with fluorescence lifetime detection on a confocal TCSPC platform.
- A single excitation laser and a broad detection window enable discrimination of FRET and non-FRET events based solely on lifetime differences.
- DNA-PAINT with fluorogenic imager strands reduces background and provides binding kinetics compatible with fast confocal scanning.
- Lifetime thresholding allows nanoscale mapping of FRET-active sites within DNA origami structures separated by sub-diffraction distances.
Why Conventional FRET Imaging Reaches Its Limits
Fluorescence resonance energy transfer is inherently a nanoscale ruler. Yet conventional FRET imaging and fluorescence lifetime imaging microscopy operate within diffraction-limited volumes. The resulting signal represents an average over many molecules that may differ in distance, orientation, or local environment. Spatial heterogeneity below ~200 nm remains unresolved.
Single-molecule localization microscopy overcomes the diffraction limit by isolating individual emission events in space and time. However, integrating fluorescence lifetime detection into SMLM is technically challenging. Camera-based localization methods lack the temporal resolution required for accurate lifetime measurements. Scanning super-resolution approaches are compatible with pulsed excitation and single-photon detectors, but lifetime analysis must remain reliable under fast acquisition conditions.
The study by Zaza et al., Small Methods (2023)1, addresses this technical gap by combining confocal scanning, time-correlated single photon counting, and DNA-PAINT into a unified workflow. The key question is whether lifetime information alone can discriminate FRET-active and non-FRET events at the single-molecule level within a super-resolved reconstruction.
Experimental Concept: Lifetime-Resolved DNA-PAINT
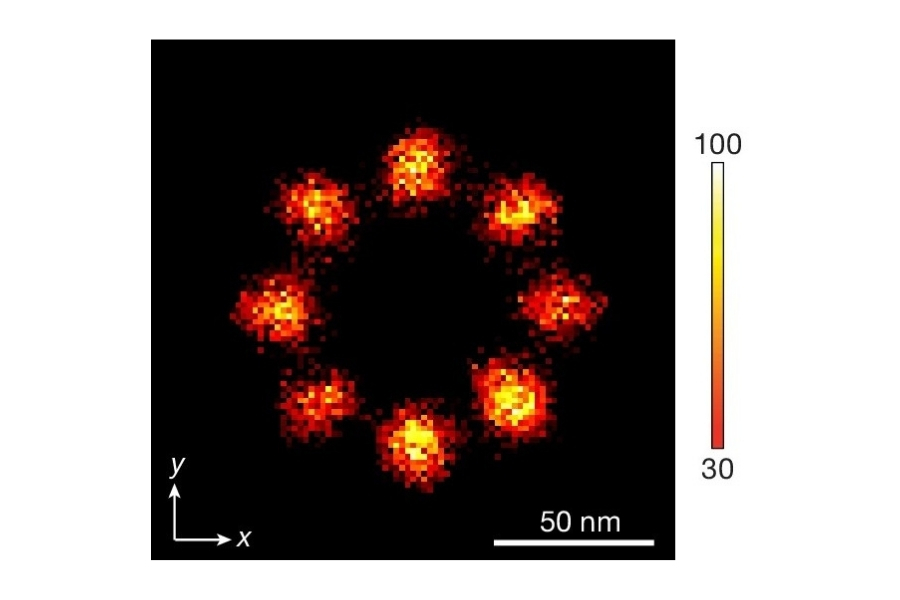
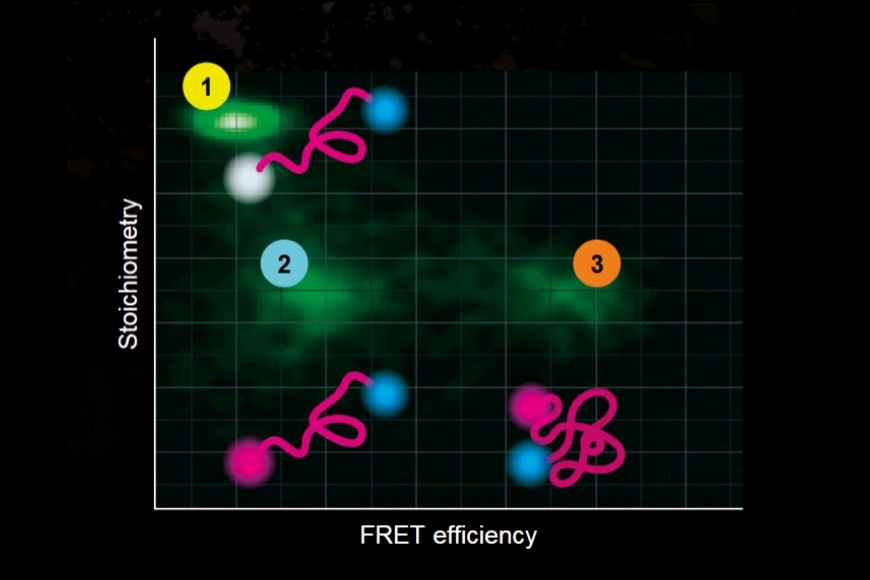
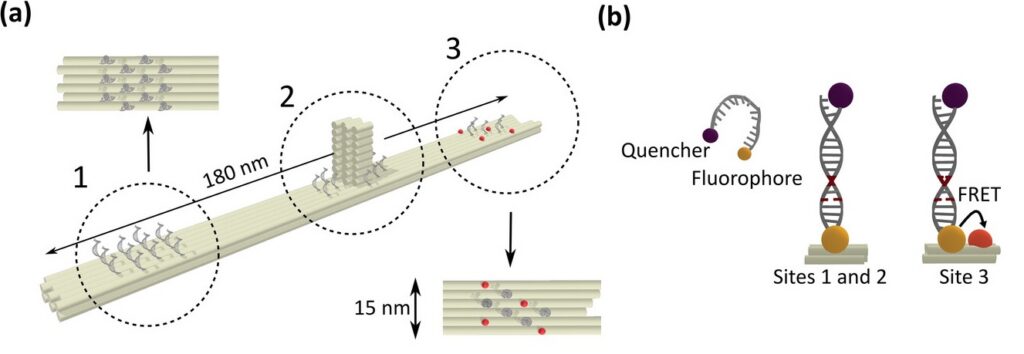
To establish a controlled nanoscale geometry, the authors employed DNA origami structures with three defined binding sites separated by 65 nm. Two sites served as donor-only references. The third site contained fixed ATTO 647N acceptors positioned within 5 to 6.3 nm of the docking strands, enabling FRET upon imager binding.
A fluorogenic DNA-PAINT imager strand carrying a Cy3B donor and a quencher was used to reduce background fluorescence. Upon hybridization, fluorescence was restored, and single-molecule binding events became detectable under confocal scanning. Excitation was performed with a pulsed 532 nm laser. Importantly, detection was not spectrally separated into donor and acceptor channels. Instead, a broad detection window collected both emissions simultaneously.
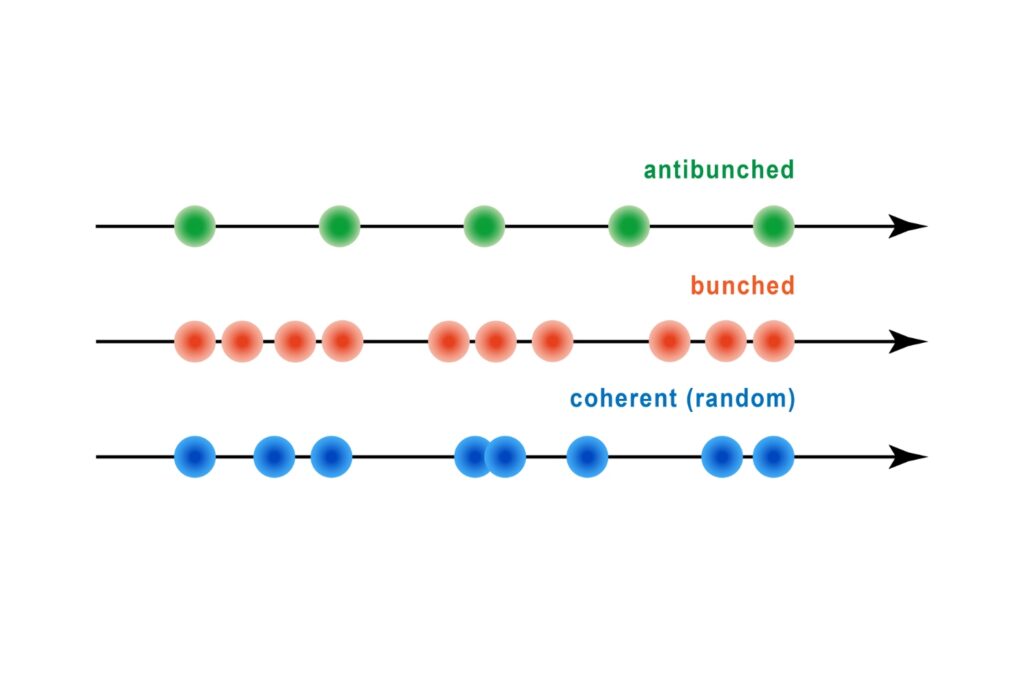
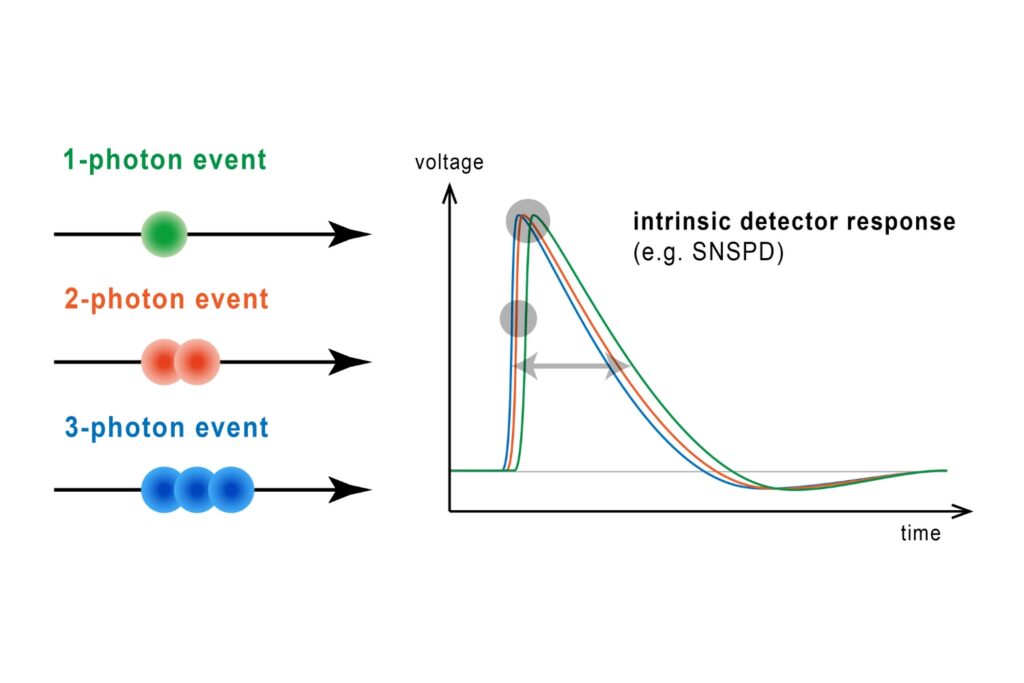
FRET discrimination relied entirely on fluorescence lifetime differences. Donor-only events exhibited shorter lifetimes around 2.7 ns, while FRET events produced a longer effective lifetime distribution due to acceptor emission contributions. This design allowed lifetime-based classification without multi-color detection.
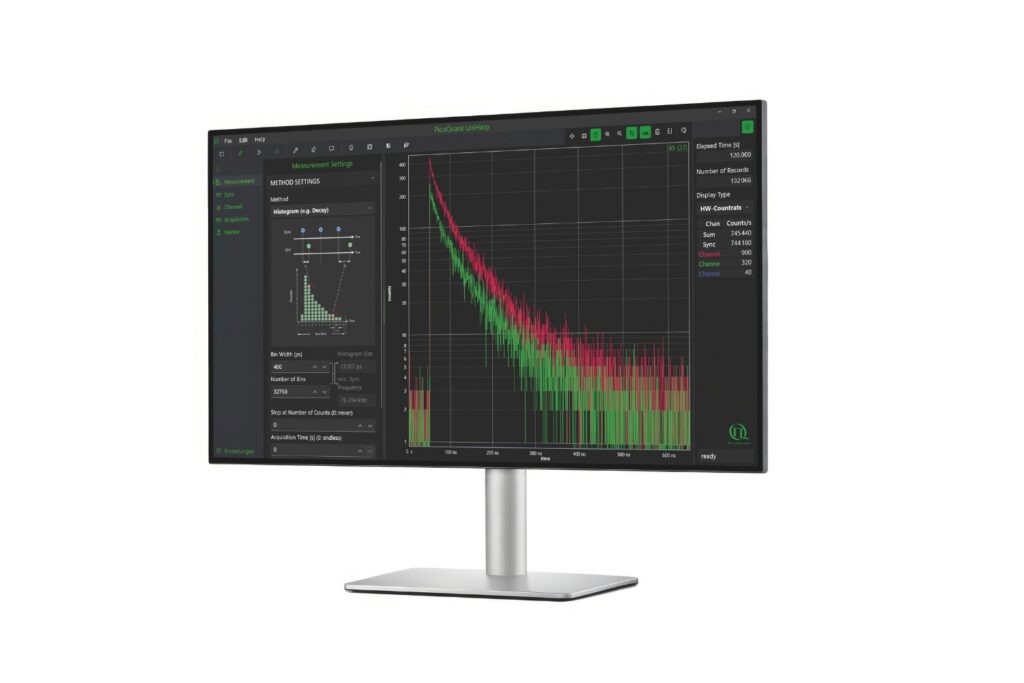
Confocal SMLM with Time-Correlated Single Photon Counting
Image acquisition required careful adaptation of scanning parameters to the binding kinetics of the imager strands. The average binding duration of 1 to 2 seconds defined the permissible acquisition window.
The confocal field of view was scanned with 60 × 60 pixels and a dwell time of 10 microseconds per pixel. Raw confocal images were acquired at 13.6 Hz. To ensure that complete single-molecule emission events were captured for localization, successive scans were binned into frames of approximately 700 milliseconds.
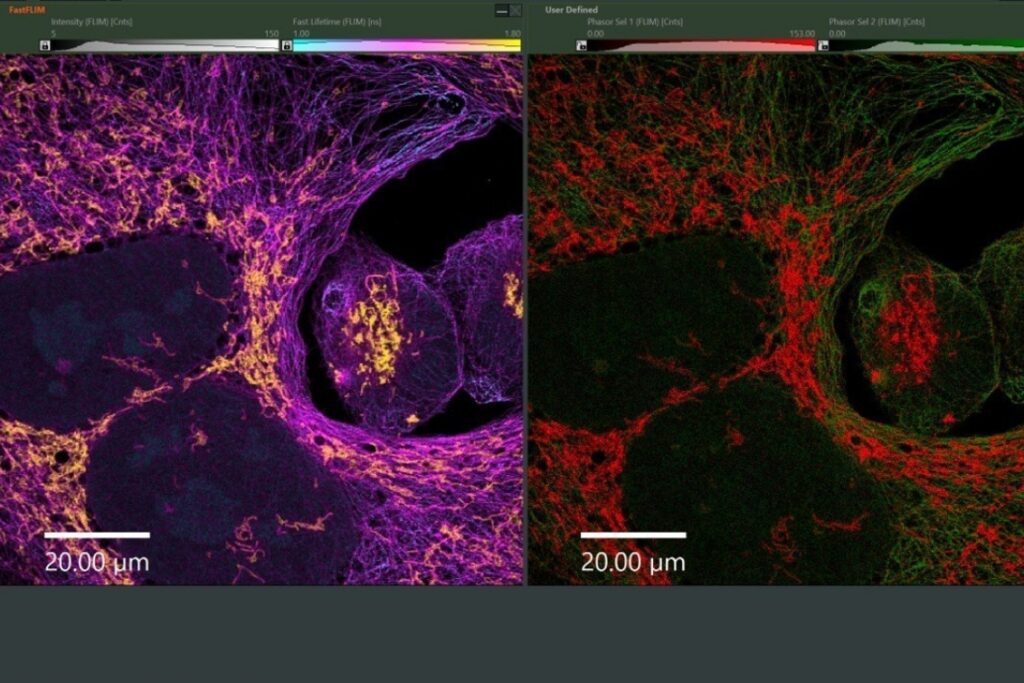
Drift correction was performed using gold nanoparticles as fiducial markers. The reconstructed confocal SMLM images resolved the three origami sites with an effective spatial resolution of approximately 20 nm. Although slightly lower than widefield camera-based SMLM in the same study, this resolution was sufficient to spatially separate FRET-active and inactive sites.
Lifetime-Based Discrimination of FRET Events
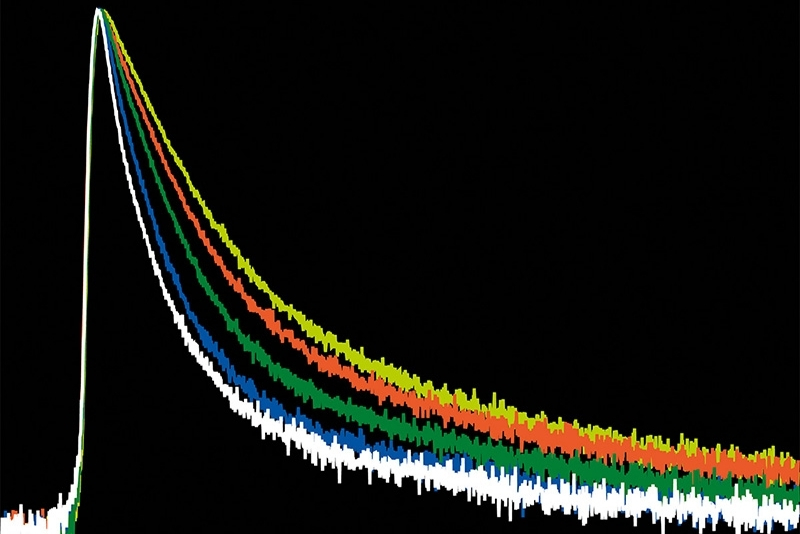
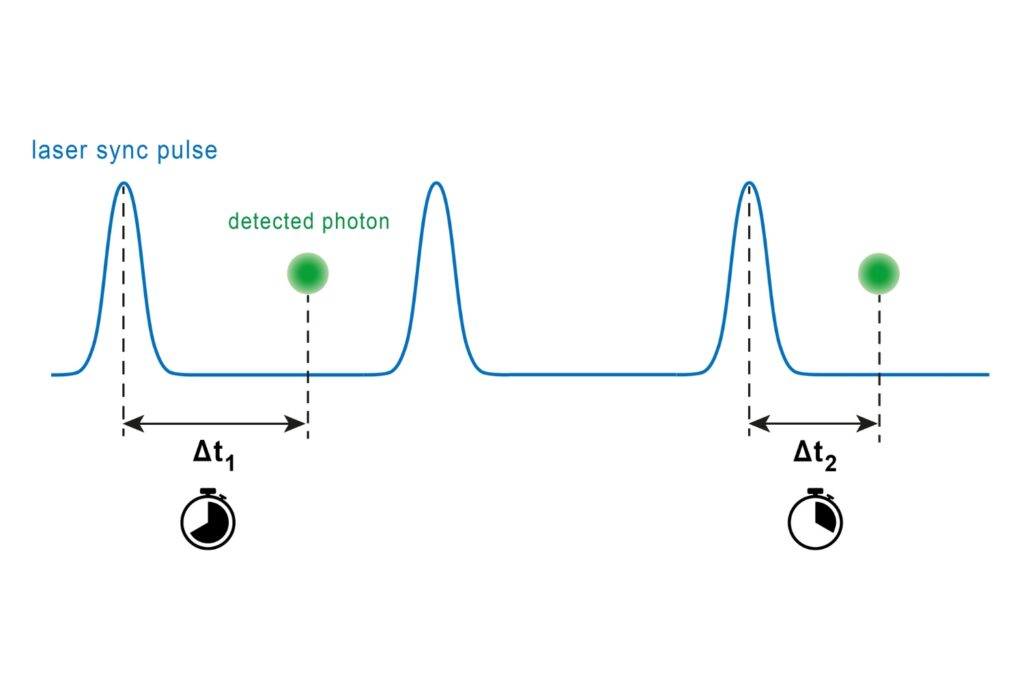
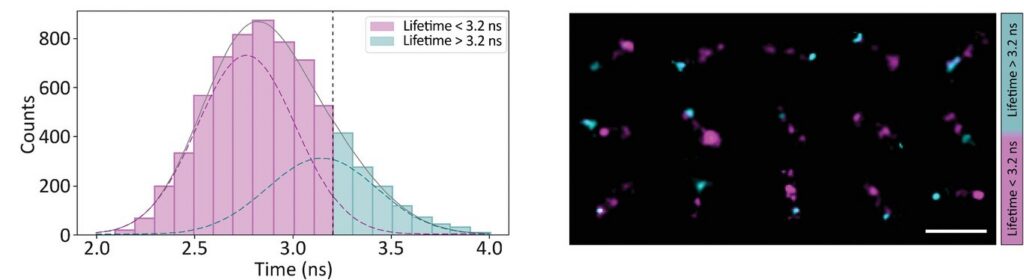
Photon arrival times associated with each localized emission event were analyzed using single-exponential fitting. The lifetime distribution of all localizations revealed two distinct populations.
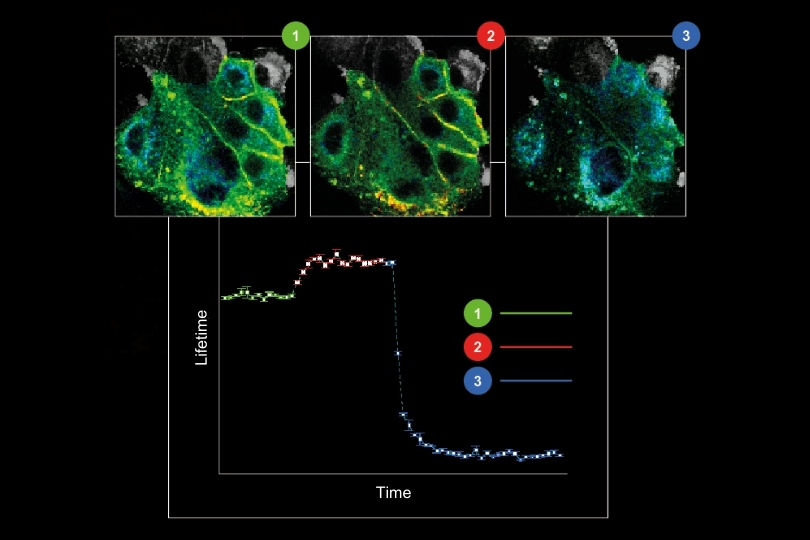
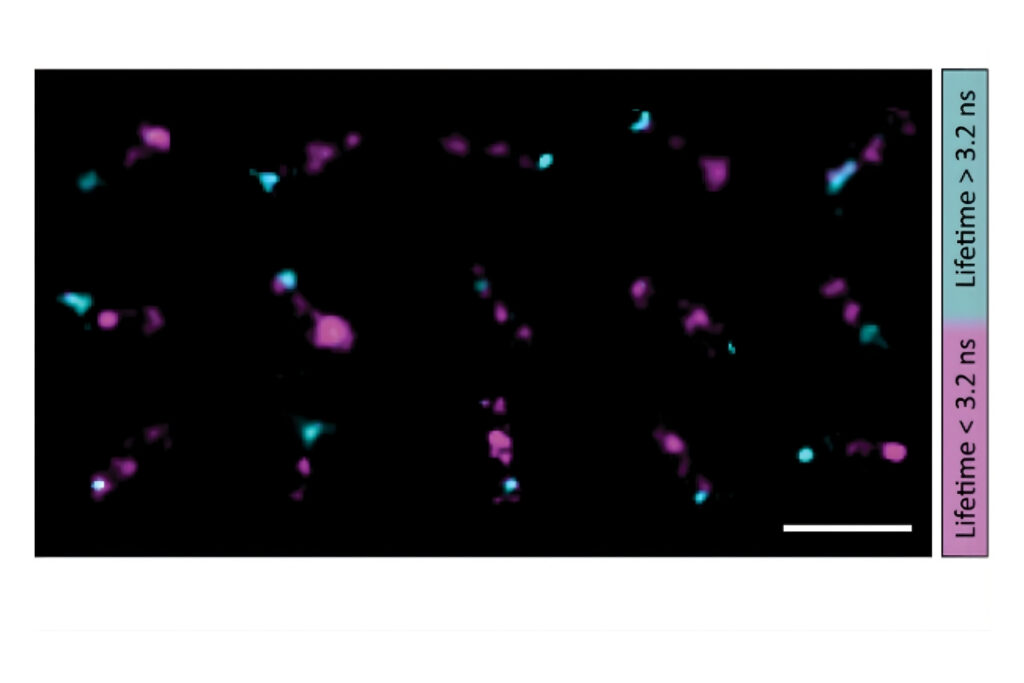
A shorter lifetime population centered at approximately 2.7 ns corresponded to donor-only emission. A longer population reflected the weighted lifetime contribution of donor emission under FRET and acceptor emission. A threshold at 3.2 ns enabled classification of single-molecule events, correctly identifying FRET occurrences with high probability.
Applying this lifetime threshold to the super-resolved reconstruction allowed nanoscale mapping of FRET-active binding sites. The FRET-capable site on each DNA origami structure was clearly identified within the super-resolution image.
Instrumentation Used in This Study by PicoQuant
Luminosa Single Photon Counting Confocal Microscope

The experiments were performed on an early prototype of the commercially available Luminosa platform. The system served as a time-resolved confocal microscope enabling single-molecule localization with fluorescence lifetime detection.
Key technical aspects relevant to this study include:
- Time-correlated single photon counting (TCSPC) integration for precise lifetime determination
- Picosecond pulsed diode laser excitation at 532 nm
- Confocal laser-scanning architecture compatible with DNA-PAINT acquisition strategiesBroad spectral detection window allowing combined donor and acceptor emission analysis
- Integration with high-performance TCSPC electronics for single-photon timing
This configuration integrates confocal FLIM and TCSPC-based detection into a single-molecule localization microscopy workflow, enabling super-resolved FRET imaging on a commercial platform.
Outlook: Toward Accessible Super-Resolved FLIM
In their conclusion, the authors emphasize that super-resolution FRET imaging can be implemented on commercial time-resolved confocal platforms. The approach does not rely on complex multi-detector synchronization or custom-built hardware. Instead, it combines established components such as pulsed excitation, TCSPC detection, and scanning microscopy within a coherent workflow.
Further improvements in drift stabilization and acquisition optimization may enhance spatial resolution to levels comparable with camera-based SMLM. The presented methodology therefore represents a technically accessible route toward integrating lifetime information into super-resolution microscopy.
1 Reference: Cecilia Zaza, Germán Chiarelli, Ludovit P. Zweifel, Mauricio Pilo-Pais, Evangelos Sisamakis, Fabio Barachati, Fernando D. Stefani, Guillermo P. Acuna. Super-Resolved FRET Imaging by Confocal Fluorescence-Lifetime Single-Molecule Localization Microscopy. Small Methods2023, 7, 2201565. https://doi.org/10.1002/smtd.202201565